
Medical device regulations are a crucial part of a country’s national health plans. They help to improve the safety and effectiveness of medical and In-Vitro Diagnostics (IVD)* devices and promote greater access to such products.
In this article, we will discuss the importance of these regulations for users and the industry, and cover some examples within the Asia-Pacific (APAC) region such as Australia, India, Japan, Singapore and South Korea. We will also share about the future of medical device regulations in the region.
*IVD devices are apparatus, instruments, equipment or other related articles used for in vitro testing of specimens derived from the human body. They can be used alone or in combination with other devices to diagnose, treat or prevent medical conditions, or in precision medicine.
- Importance of Medical Device Regulations
- Examples of Medical Device Regulations in APAC Countries
- The Future of Medical Device Regulations in APAC
- FAQs About Medical Device Regulations
Importance of Medical Device Regulations
Implementing regulations on medical devices safeguards the interests of users, promotes greater vigilance, and encourages growth and greater compliance within the industry.
Ensures users’ safety
Medical devices are created to enhance the lives of users and improve their health and wellbeing. However, certain products such as pacemakers and implants may pose a relatively high safety and health risk to users.
To ensure their safety, rules governing these devices are implemented and higher-risk products are often subject to tighter regulations.
Promotes greater compliance and standardization across the industry
Medical device regulations typically outline the basic standards and requirements for the development and manufacturing of such devices. This ensures greater compliance from manufacturers, promotes standardization across the industry and improves quality control.
Promotes greater vigilance
These regulations promote greater vigilance among users and manufacturers alike as they are encouraged to report the occurrence of adverse events. This enables corrective action to be taken as quickly as possible.
Moreover, manufacturers are required to collect performance data regarding devices, allowing for early detection of potential safety hazards.
Improves the innovation process
Providing a standardized and accessible set of regulations that govern the industry allows manufacturers to create products that adhere to the requirements of the exporting country.
This improves the efficiency of the innovation process, encourages the production of safer devices, and reduces the overall cost for users.
Examples of Medical Device Regulations in APAC Countries
Medical device regulations tend to vary across different regions and countries with regard to their scope, risk classification and registration pathways.
Here are examples of regulations implemented across countries within the APAC region, namely Australia, India, Japan, Singapore and South Korea.
Medical device regulations in Australia

In Australia, the governing body for medical devices is the Therapeutic Goods Administration (TGA).
It assesses and monitors various therapeutic goods to ensure that they are of an acceptable standard to be sold and used within the country.
Scope
All medical devices, including IVD devices, are subject to the following regulations:
- The Therapeutic Goods Act 1989 (the Act)
- The Therapeutic Goods Regulations 1990
- The Therapeutic Goods (Medical Devices) Regulations 2002 (the Regulations)
For any such device to be imported into, supplied within, or exported from Australia, it must be registered in the Australian Register of Therapeutic Goods (ARTG), unless stated otherwise under the Act.
Risk classification
These are the main risk classes for medical devices in Australia. They are classified based on their potential risk to users:
| Risk class | Risk level | Examples of medical devices |
| I | Lowest | |
| Is, Im, IIa | Low to medium | |
| IIb | Medium to high | |
| III | High |
Registration pathways
Before manufacturers begin registering their devices in the ARTG, they must first determine if registration is necessary. If it is necessary, they will need to provide information such as:
- Type of medical device
- Manufacturer’s evidence
- Risk classification
After submitting their application based on the risk classification and obtaining approval, they can print their ARTG certificate of inclusion.
Medical device regulations in India

Medical devices, along with drugs, biologics, and cosmetics, are regulated by the Central Drugs Standard Control Organization (CDSCO).
It falls under the purview of the Ministry of Health and Family Welfare through the Directorate General of Health Services (DGHS) headed by the Drugs Controller General of India (the DCGI).
Scope
Registration procedures for all types of medical devices regulated under the Medical Device Rules are implemented in phases. They have been classified following a risk-based approach in accordance with MDR-2017 Chapter II, Rule4(3).
The CDSCO has classified the non-notified medical devices based on the intended use, associated risks, and other parameters specific to the device. The classification list is dynamic and subject to changes.
Risk classification
Under the Medical Device Rules 2017, medical devices and in-vitro diagnostic devices are classified based on their intended use and associated risks. They can be classified into the following categories:
| Risk class | Risk level | Examples of medical devices |
| A | Low | |
| B | Low-moderate | |
| C | Moderate-high | |
| D | High |
A list of classifications for specific products can be found in DCGI’s Notice regarding Classification of Medical Devices and In Vitro Diagnostic Medical Devices.
Registration pathways
Manufacturers can register their devices in India via their online portal. They will require certain information and documents for the registration such as:
- Name and address of the company or entity:
- Manufacturing the medical device along with name and address of the device’s manufacturing site or
- Importing the medical device
- Email address
- Phone number
- Medical device details (e.g. dimensions, materials, risk class and shelf life)
- Certificate of Compliance (e.g. ISO 13485)
- Undertaking signed by the manufacturer regarding the authenticity of the information
The full list of information and documents required can be found here. Alternatively, you may also refer to the Fourth Schedule of Medical Device Rule 2017.
Medical device regulations in Japan

The Pharmaceutical and Medical Device Agency (PMDA) and Ministry of Health, Labor, and Welfare (MHLW) work together to regulate and assess pharmaceutical products and medical devices in Japan.
Scope
All medical devices and software are regulated under the PMD Act, which covers areas such as:
- Product registration
- Foreign Manufacturer Registration (FMR)*
- Quality management system compliance
- Third-party certifications
*This is mandatory for foreign manufacturers whose devices are produced outside of Japan but are to be sold and marketed within the country.
Risk classification
The Japan Medical Device Nomenclature (JMDN) system segments medical devices into 4 risk classes based on their potential risk.
| Risk class | Risk level | Requires approval/ certification from | Examples of medical devices |
| I: General medical device | Low | Approval/certification not required. Only requires a notification/ self-declaration. | |
| II: Controlled medical device | Moderately low | A Registered Certification Body (RCB) or PMDA | |
| III: Specially controlled medical device | High | PMDA and MHLW | |
| IV: Specially controlled medical device | Very high (life-threatening) | PMDA and MHLW |
Registration pathways
Depending on the risk class, manufacturers can follow 3 different pathways for registering their devices:
- Pre-market submission (Todokede)
This registration pathway is applicable to Class I devices, which do not require a review or assessment by the PMDA. Only notifications are required and this can be done via a pre-market submission.
- Pre-market certification (Ninsho)
Class II and certain Class III devices, which have a specific certification standard, require pre-market classification.
In this registration pathway, the Marketing Authorization Holder (MAH) or in-country representative will submit an application to PMDA or a Registered Certification Body (RCB).
- Pre-market approval (Shonin)
Pre-market approval is required for Class IV devices and Class II and III devices that do not have a specific certification standard. In this process, the MAH will apply with the PMDA for device registration and obtain approval from the MHLW.
Medical device regulations in Singapore

In Singapore, all medical devices are regulated by the Health Sciences Authority (HSA).
Scope
A dealer’s license is required for companies that seek to manufacture, import or sell medical devices within the country. These devices are subject to the Health Products Act (HPA) and the Health Products (Medical Devices) Regulations 2010.
The HSA also requires companies to comply with regulations related to the
- notification of changes for registered devices (e.g. technical or administrative changes),
- advertisement and promotion of devices,
- reporting of adverse events, and
- reporting of Field Safety Corrective Actions (FSCA).
Risk classification
There are 4 main risk classification categories – A, B, C and D.
| Risk class | Risk level | Examples of medical devices |
| A | Low | |
| B | Low-moderate | |
| C | Moderate-high | |
| D | High |
Registration pathways
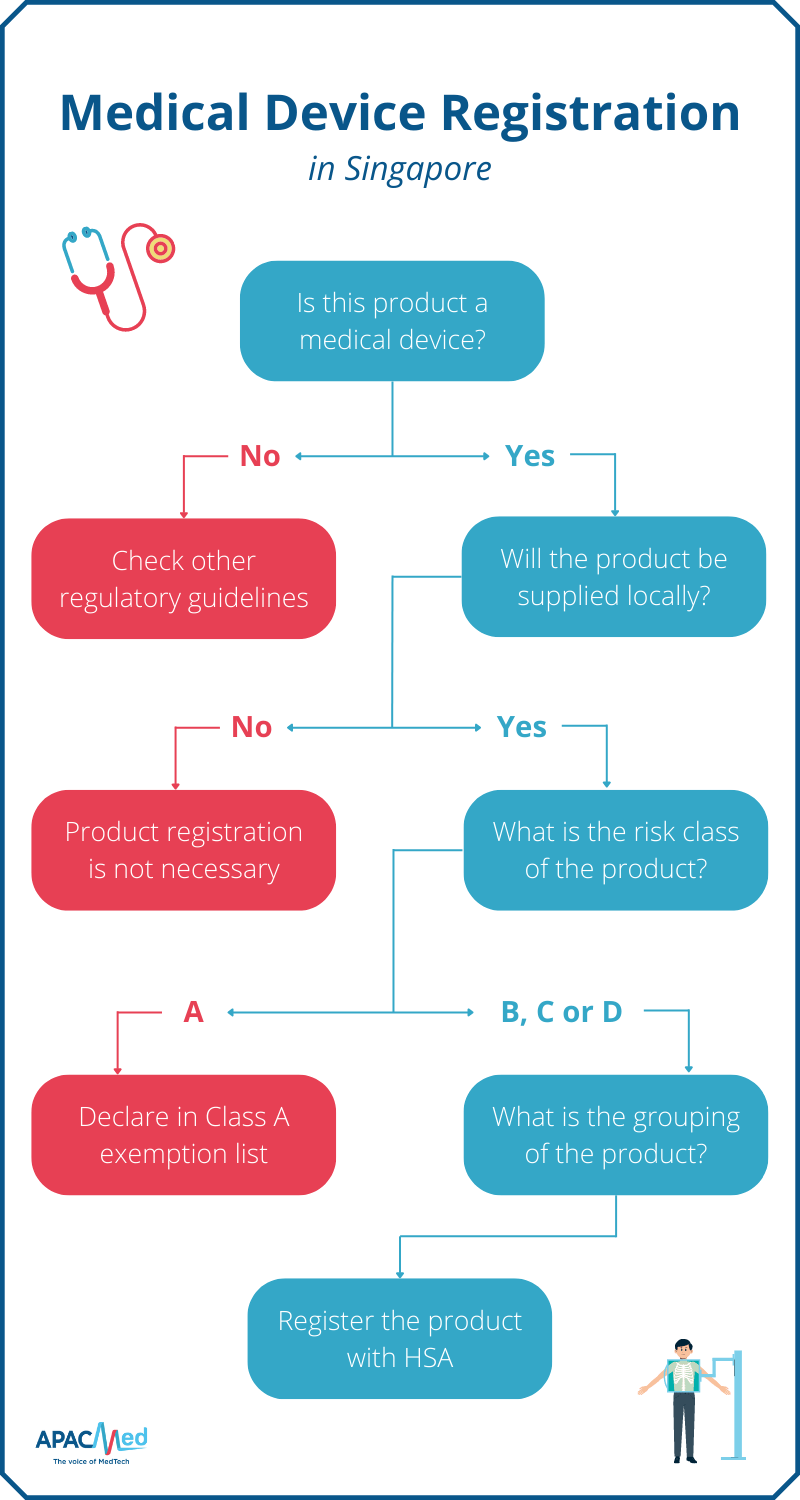
Manufacturers can start by determining if the product is considered as a medical device based on HSA’s definition. All medical devices, except Class A products and those exempted by HSA, must be registered before they can be sold and supplied in Singapore.
If the device is to be re-exported, product registration is not necessary. If the device falls into the Class A category, manufacturers can declare it under the Class A exemption list – registration is also not required.
Once the product is confirmed to be a medical device that requires registration with HSA, manufacturers are recommended to determine the grouping of the device before proceeding with the application.
Medical device regulations in South Korea

The governing body responsible for the regulation of medical devices in South Korea is the Ministry of Food and Drug Safety (MFDS).
The Medical Device Information and Technology Assistance Center (MDITAC) and the National Institute of Medical Device Safety Information (NIDS) will also assist with the certification of certain classes of devices.
Scope
Within South Korea, this group of products is regulated based on the Medical Device Act (MDA). A total of 2,334 medical devices*, including 225 IVD devices, are recognized under the country’s regulations.
*Accurate as of the date of publication.
Risk classification
In South Korea, medical devices can be classified into 4 risk classes:
| Risk class | Risk level | Type of application | Examples of medical devices |
| I | Very low | Notification | |
| II | Low | Certification or approval | |
| III | Moderate | Approval | |
| IV | High | Approval |
Registration pathways
The registration pathways vary according to the risk class of the medical device. Do note that all documents are to be submitted in Korean.
There are 3 main processes – pre-market notification, classification and approval:
- Pre-market notification
Pre-market notification is applicable for Class I devices and is to be filed with the MDITAC and NIDS.
- Pre-market certification
Some Class II devices require pre-market certification from the MDITAC and NIDS.
- Pre-market approval
Other Class II and all Class III and IV devices will go through the pre-market approval process with the MFDS.
A General Technical File Review or Technical Safety & Efficacy Review (SER) is required for this process. While clinical study reports are not mandatory for the General Technical File Review, they are necessary for Technical SERs.
The Future of Medical Device Regulations in APAC
Some of the common issues faced within the APAC region as observed by Elisabeth Staudinger from Siemens Healthineers are rising costs, a growing aging population, and unequal access to medical care.
With more regional collaborations and greater regulatory convergence with regard to medical devices, manufacturers can strive toward producing safer, more effective and less costly products for the region. Patients can also enjoy greater access to such products.
As such, one of the goals of APACMed is to advocate for policies and regulations that promote access to innovative and beneficial medical technologies, especially amidst healthcare digitization.
Greater regulatory convergence
To achieve greater regulatory convergence within the region, APACMed strives to build long-term partnerships and share best practices with relevant stakeholders such as government agencies and industry associations.
An example of the organization’s efforts includes an exploration of regional reliance protocols for ASEAN countries. Through this set of protocols, APACMed seeks to speed up the process of medical device approvals and increase patients’ access to such products.
Increased number of regional collaborations
- Singapore and Thailand
According to Anirudh Sen from APACMed, the organization has been working with various regulatory agencies in the region to initiate pilot reliance programs.
An example of this is the collaboration with the HSA of Singapore and the Food and Drug Administration (FDA) of Thailand to expedite the registration process for Class B, C and D medical and IVD devices through regulatory reliance pathways.
As part of this program, the Thailand regulatory body will assess the devices based on the safety assessment and performance results from the HSA.
- Philippines and ASEAN countries
In the Philippines, a program has been implemented to encourage greater regulatory reliance within the ASEAN region. This collaboration takes place together with Singapore, Thailand, Indonesia and Malaysia, and is achieved through abridged pathways for Common Submission Dossier Template (CSDT) document submission.
This program is applicable to Class B, C, and D medical devices and helps to promote a quicker approval process.
FAQs About Medical Device Regulations
What are medical devices?
The World Health Organization (WHO) defines medical devices as instruments, appliances, machines or other related articles intended for medical use, whether alone or in conjunction with other devices.
Some examples of these devices include wearable ECG devices, blood pressure monitoring devices, X-ray machines, heart valves and implantable defibrillators.
Do medical device regulations include IVD devices?
Yes, most, if not all, medical device regulations cover IVD products. It is important to note, however, that the regulations for these products may differ slightly from those of general medical devices.
What is missing from the roadmap in terms of medical device regulation?
According to Anirudh Sen from APACMed, a harmonized regulatory process is needed within the ASEAN region.
Currently, medical device regulations differ across countries in the region, and this can potentially slow down the application and approval process. This may, in turn, limit patients’ access to such technologies.


